


Copyright © 2025 Shenyang Dasan Pharmaceutical Technology Co., Ltd.

 Posting Date:2023-05-19
Posting Date:2023-05-19 Views:
Views: Reversed-phase bonded phase chromatography (C18, C8 stationary phases) is currently one of the most commonly used chromatographic methods. When it is employed for pharmaceutical impurity testing, especially for impurities with low response factors, a large injection volume is often used to enhance the method's detection capability for impurities. In such cases, overloading of the column with the main component often occurs.
According to chromatographic kinetics, overloading is caused by non-linear isothermal distribution. Chromatographic theory holds that components are distributed proportionally between the stationary phase and the mobile phase. When the stationary and mobile phases are fixed, the distribution coefficient is only a function of temperature. When the amount of a component is within a certain range, the distribution coefficient remains constant, and the amount of the component in the stationary phase (MS) and mobile phase (Mm) follows a linear relationship. When the amount of the component exceeds this range, the distribution deviates from linearity (non-linear isothermal distribution), resulting in column overload. Overload manifests in two ways:
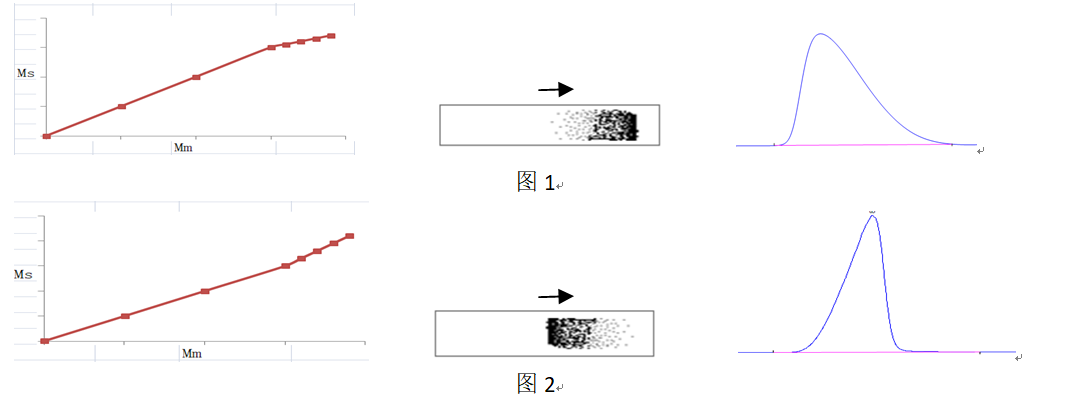
The first scenario, as shown in Figure 1, involves a decrease in k’ as the component amount increases after column overload. On the chromatographic band, this manifests as faster migration in regions of higher concentration, resulting in a chromatographic peak with an approximately right-triangular shape (overload tailing).
The second scenario, as shown in Figure 2, involves an increase in k’ as the component amount increases after column overload. On the chromatographic band, this manifests as slower migration in regions of higher concentration, resulting in a chromatographic peak with an approximately right-triangular leading edge (fronting).
The first scenario (overload tailing) is very common in HPLC. The following discussion primarily focuses on overload tailing.
1. Characteristics and Adverse Effects of Overload Tailing Peak Shape
Typically, it appears as an approximately right-triangular shape. The larger the injection volume, the more severe the tailing and the shorter the retention time. Although retention times vary with different injection volumes, the peaks all end at the same time.
Since the peak end position remains unchanged, such tailing peaks do not affect the separation of impurities eluting after them, but they can easily overlap with impurity peaks eluting before them, which is detrimental to their separation.
2. Factors Influencing Overload and Solutions
(1) Column Capacity
Higher column capacity makes overloading less likely. The indicator of column capacity is the surface area ratio of the stationary phase on the column cross-section, which is related to chromatographic parameters such as void volume, specific surface area of the packing material, and surface bonding density.
It is important to note that increasing column length does not increase column capacity because the sample cannot be immediately distributed throughout the entire column after injection. Similarly, due to the infinite diameter effect, increasing the column internal diameter does not increase column capacity.
(2) Injection Volume
Obviously, reducing the injection volume can alleviate overloading and sometimes achieve separation of impurities.
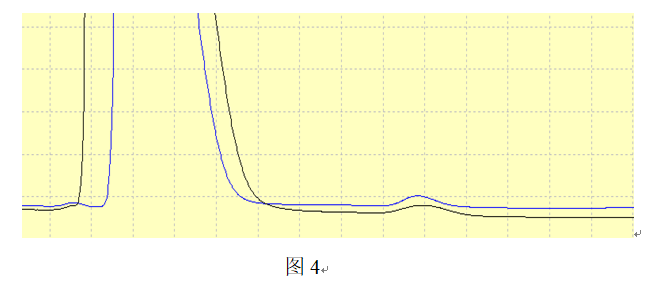
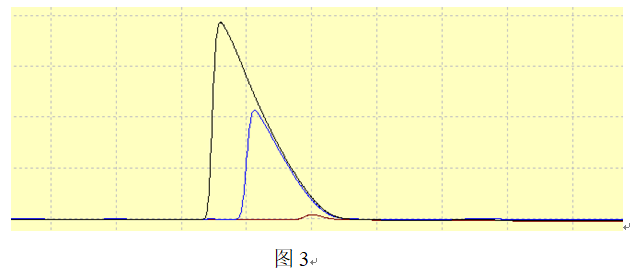
As shown in Figure 4, when the injection volume is halved, an impurity peak adjacent to the front of the main component is separated from the main component peak, but the method's ability to detect impurities is correspondingly reduced.
(3) Amount of Component Distributed to the Stationary Phase
When column capacity is fixed, the less the component distributes into the stationary phase, the lower the retention, and the less likely the column is to be overloaded. Theoretically, if a component does not distribute into the stationary phase, it will not be retained, and even large injection volumes will not cause overloading. For C18/C8 bonded stationary phases, overloading can be alleviated by increasing the organic phase proportion to reduce the distribution of the component in the stationary phase. Of course, method selectivity may also change with the organic phase composition.
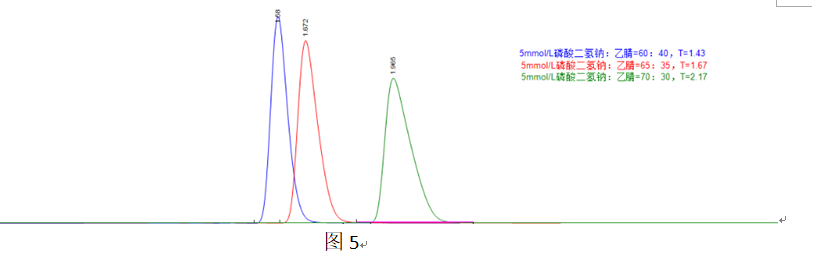
In Figure 5, increasing the organic phase proportion reduces retention, alleviates overloading, and decreases the tailing factor.
(4) Existing Form of the Component
For components that readily dissociate, such as acidic or basic substances, under conditions where the buffer capacity of the mobile phase is sufficient, the ratio of dissociated (ionic) to non-dissociated (molecular) forms at any point on the band remains constant. The ionic and molecular forms flow out of the column together at a constant ratio, and the resulting chromatographic peak is the sum of the ionic and molecular forms.
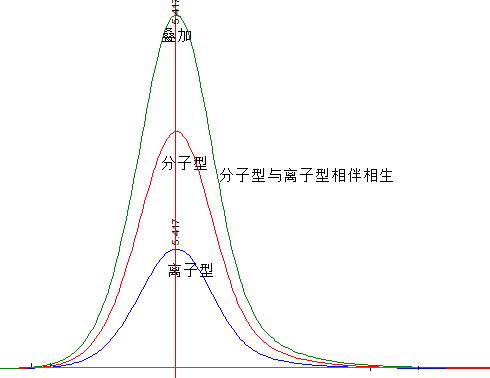
Figure6
If one component form is overloaded, exhibiting a tailing chromatographic peak, the other component form, although not overloaded, will also appear as an overload peak of similar shape but different magnitude due to the aforementioned reasons. Therefore, the peak shape of a readily dissociable component is determined by the overloaded existing form.
Generally, the ionic form is more prone to overloading than the molecular form. Hence, overloading of a readily dissociable component is determined by the overloading of the ionic form, and the degree of overloading is related to its proportion. Adjusting the mobile phase pH to suppress component dissociation can significantly reduce the proportion of the ionic form, alleviating or even eliminating overloading.
For strong acid/base substances, it is difficult to suppress dissociation by adjusting pH. In such cases, the proportion of the ionic form can be reduced by forming ion-pair complexes to alleviate overloading. Similarly, overloading is determined by the ionic form; the more ion-pair complexes formed, the less the ionic form, and the more overloading is alleviated.
A more complex scenario involves both dissociation suppression and the presence of ion-pair reagents. In this case, the proportion of the ionic form can be altered through the two aspects mentioned above, which can alleviate overload tailing and also adjust retention. Although the situation is complex, the approaches are more flexible and diverse.
(5) Salt Concentration
The reason ionic components readily overload on C18/C8 stationary phases is primarily due to electrostatic repulsion between ionic species, which hinders further entry of components once they enter the stationary phase.
Buffer salts are typically used mainly to maintain sufficient buffer capacity in the mobile phase. However, buffer salts, and even neutral salts, can also increase the ionic strength of the mobile phase, partially shielding the electrostatic repulsion between ionic component molecules, thus reducing overloading. Additionally, salts can form ion-pair complexes with ionic components, reducing the proportion of the ionic form and alleviating overloading. Higher salt concentrations yield more pronounced effects.
Using a specific basic compound as an example, the following experiments investigate the effect of salt on overload behavior. This compound contains a strongly hydrophobic aromatic group and a primary amino group, is strongly basic with a pKa of 9.9, and exists almost entirely in dissociated form at the mobile phase pH used in the experiments. For all experiments below, the buffer pH was adjusted to 2.5 with phosphoric acid; the organic phase in the mobile phase was acetonitrile; except for the acetonitrile proportion indicated in the figures, the mobile phase composition was buffer: acetonitrile = 80:20; column temperature: 30°C; detection wavelength: 210 nm; column: Agilent SB-C18 (4.6*150mm, 5um); sample concentration: 1 mg/ml; injection volume: 10 uL.
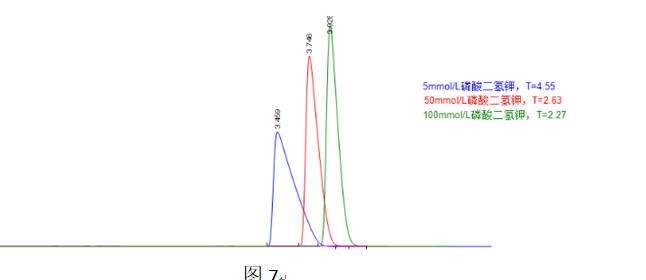
Figure 7 shows that as the buffer salt concentration increases, overloading is alleviated, and the peak tailing factor gradually decreases. Even the addition of neutral salt like sodium sulfate produces a similar effect.
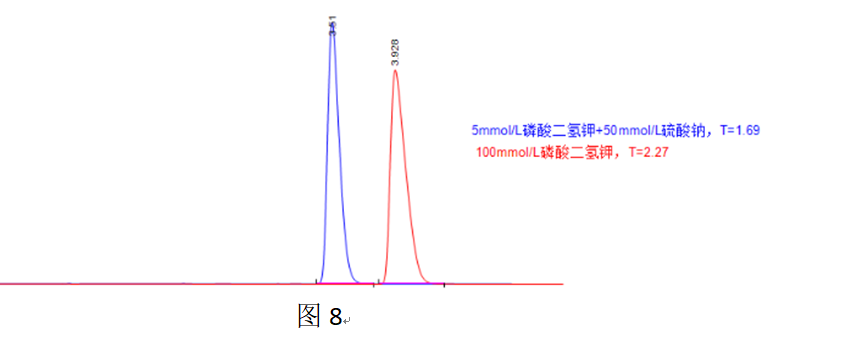
In Figure 8, 50 mmol/L sodium sulfate alleviates overload tailing more effectively than 100 mmol/L potassium dihydrogen phosphate, which is attributed to the higher ionic strength of 50 mmol/L sodium sulfate.
In Figure 7, the three chromatographic peaks do not end at the same time, indicating a change in the capacity factor with increasing buffer salt concentration. The figure shows that higher salt concentration leads to a larger capacity factor, suggesting the formation of more strongly retained ion-pair complexes between the component and the dihydrogen phosphate anions from the buffer salt. One puzzling observation is that the capacity factor did not increase as expected when using 50 mmol/L sodium sulfate in Figure 7.
The proportion of ion-pair complexes formed between the buffer salt and the ionic component has always been a point of interest. This proportion can be calculated using the following formula:
k’=xionic﹒k’ionic +xion-pair﹒k’ion-pair
x represents the proportion of each existing form, and k’ is the corresponding capacity factor.
The retention of the ion-pair complex should be comparable to that of the molecular form. Considering the strongly basic nature of the component, to determine the retention value of the component's molecular form, it is necessary to completely suppress its dissociation, which would require a mobile phase pH above 12. This is difficult to achieve on a conventional C18 column. Therefore, a compound where the primary amino group is replaced by an amide group was used as a substitute for the molecular form of the target substance. The retention value measured for this substitute was used as the retention value for the ion-pair complex (k’ion-pair) in the calculation.
k’ionic was approximated using the k’ value obtained at the lowest buffer salt concentration (5 mmol/L).
The chromatographic peaks of the component at various buffer concentrations all exhibited some degree of overload tailing. Therefore, retention times were estimated using the peak end times, which should not introduce significant error (ideally, the component should be injected at a low concentration under each condition to obtain an accurate retention value without overloading).
|
Buffer Concentration (mmol/L) |
t0(min) |
tR(min) |
K’ |
xion-pair |
|
5 |
1.22 |
3.70 |
2.03 |
0 |
|
50 |
3.83 |
2.14 |
0.0026 |
|
|
100 |
3.95 |
2.24 |
0.0049 |
|
|
Amide-substituted |
55.8 |
44.7 |
— |
Thus, even at a buffer salt concentration as high as 100 mmol/L, the proportion of ion-pair complexes formed between the dihydrogen phosphate anion from the buffer salt and the cationic component is less than 1%.
Generally, the stronger the ion polarization ability and the larger the hydrated ionic radius, the greater the ability to form ion-pair complexes. PF6— and ClO4— are among the most effective anionic ion-pair reagents due to their strong polarizing ability and large hydrated ionic radius. Typically, the order of ion-pair forming ability is PF6— > ClO4— >> other anions.
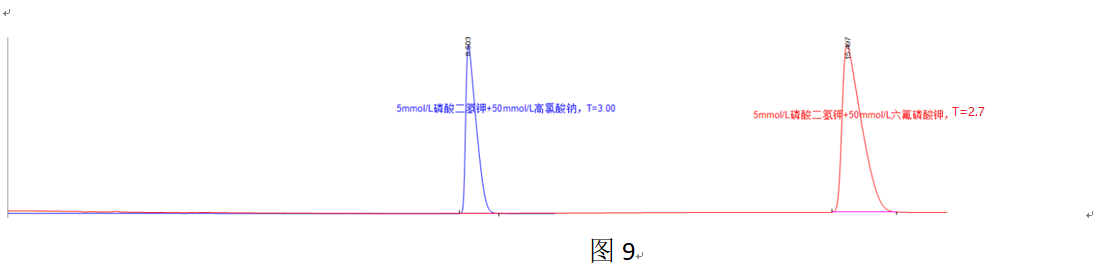
Figure 9 shows that using potassium hexafluorophosphate results in stronger retention compared to using the same concentration of sodium perchlorate, indicating that PF6— has a greater ability to form ion-pair complexes than ClO4—. Consequently, the former alleviates overloading to a greater extent than the latter, resulting in a lower tailing factor.

In Figure 10, the retention value at 55.8 min corresponds to the amide-substituted compound, used as a substitute for the ion-pair complex retention. Using this, the calculated proportions of ion-pair complexes formed with the component in 50 mmol/L sodium perchlorate and 50 mmol/L potassium hexafluorophosphate are 9.2% and 22.7%, respectively.
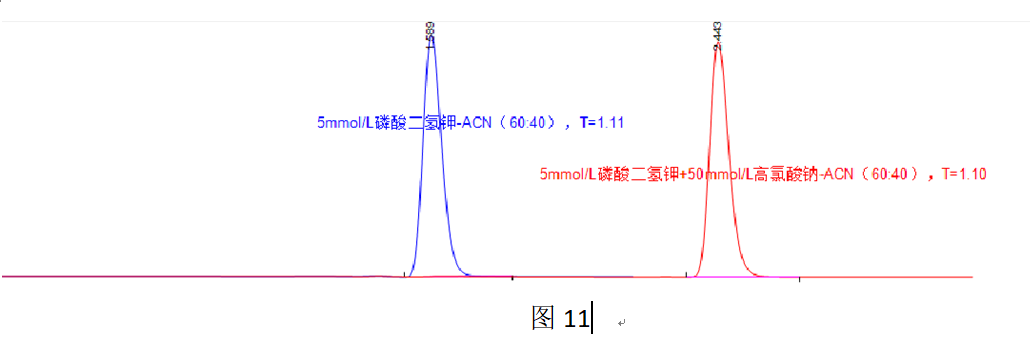
Since overloading is caused by the ionic form, increasing the acetonitrile proportion in the mobile phase can reduce the distribution of the ionic form in the stationary phase, thereby mitigating or even eliminating overloading. Figure 11 shows that when the acetonitrile proportion in the mobile phase is increased to 40%, the component, which exists almost entirely in the ionic form, hardly distributes into the stationary phase, and the chromatogram shows no signs of overloading (T = 1.11).
However, non-distribution (or low distribution) also implies non-retention (or low retention). To achieve adequate retention of the main component, ion-pair reagents can be added. In Figure 11, the addition of 50 mmol/L sodium perchlorate enables component retention, while the chromatographic peak tailing factor remains consistent with the almost non-retained condition, further demonstrating that overloading is caused by the ionic form, and alleviating the overloading of the ionic form alleviates the overloading of the entire chromatographic peak.
The method used above involves using a high acetonitrile proportion to prevent the ionic component from distributing into the stationary phase, thereby eliminating overload tailing, and then adding an ion-pair reagent like sodium perchlorate to achieve appropriate component retention. To achieve even stronger retention, hydrophobic ion-pair reagents with different alkyl chains, such as alkyl sulfonates/sulfates (e.g., sodium heptanesulfonate), can be used. These types of ion-pair reagents have a weak ability to form ion-pair complexes and are typically used at very low concentrations (a few tenths to several mmol/L). Consequently, the concentration of ion-pair complexes formed is very low, making it difficult to effectively reduce the proportion of the ionic component, and they contribute little to alleviating overloading. However, due to their long carbon chains, the few ion-pair complexes that do form have a k’ that can be much larger than that of the molecular component, effectively prolonging component retention.
Regarding the retention mechanism of such ion-pair reagents, compared to sodium perchlorate and potassium hexafluorophosphate, due to their strong hydrophobicity, in addition to the ion-pair complex formation mentioned above, there is also a so-called “dynamic ion exchange” retention mode. In this mode, the ion-pair reagent distributes onto the C18 stationary phase, forming a dynamic stationary phase with cation exchange functional groups, which exerts a retention effect similar to ion exchange for cationic components. The effective column capacity for this ion exchange is related to the organic phase proportion in the mobile phase, as there is competition between the organic phase and the ion-pair reagent for distribution onto the stationary phase. The higher the organic phase proportion, the less ion-pair reagent distributes onto the C18 phase, and the lower the effective column capacity for ion exchange.
Overloading with such ion-pair reagents involves not only overloading of the ionic form on the C18 phase but also overloading of the ionic form on the so-called “ion-exchange stationary phase”. A simple and effective way to alleviate or eliminate these two types of overloading is to use a high organic phase proportion in the mobile phase to reduce the distribution of the ionic component on the C18 stationary phase, while simultaneously using a high salt concentration to reduce the distribution of the ionic component on the “ion-exchange stationary phase.”
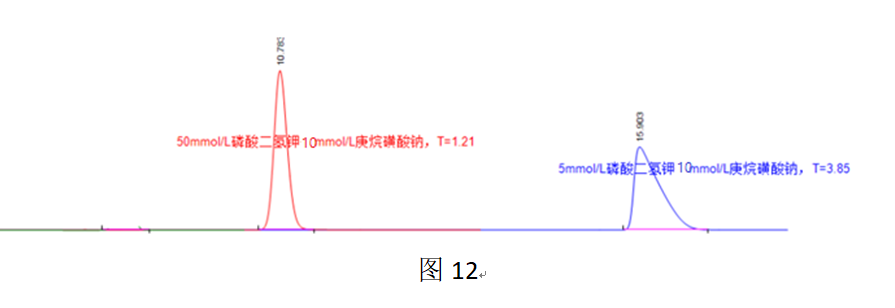
In Figure 12, both chromatographic peaks were obtained using a mobile phase with 40% acetonitrile, where overloading of the ionic component on the C18 stationary phase is essentially eliminated. At a mobile phase salt concentration of 5 mmol/L, overload tailing is evident, attributed to overloading of the ionic component on the “ion-exchange stationary phase.” When the mobile phase salt concentration is increased to 50 mmol/L, the tailing is significantly alleviated.
The use of ion-pair reagents with longer carbon chains has always been problematic! For instance, sodium dodecyl sulfate can easily lead to distorted peak shapes.
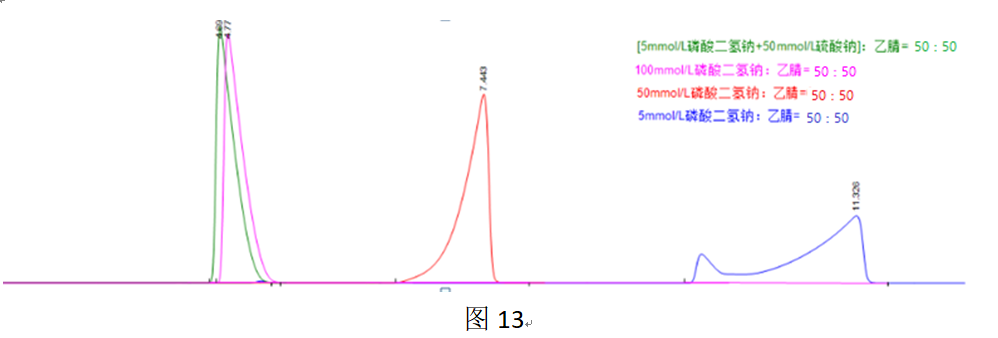
In Figure 13, the mobile phase contained 2.5 mmol/L sodium dodecyl sulfate. A normal peak shape was only achieved when the buffer salt concentration was as high as 100 mmol/L. The reason for this might be that the distribution of long-chain ion-pair reagents on the C18 phase can cause orientation disorder, leading to an uneven distribution of the sulfate groups forming the “ion-exchange stationary phase” on the C18 phase. Increasing the salt concentration in the mobile phase can weaken or even eliminate the ion-exchange effect, thereby reducing or eliminating the retention effect of this inhomogeneous “ion-exchange stationary phase.”
(6) Gradient Elution
During gradient elution, as the organic phase proportion in the mobile phase gradually increases, the distribution of the component in the stationary phase also gradually decreases, thus progressively mitigating the accumulation of overloading. Furthermore, even if overloading occurs early during elution with a low organic phase proportion, the unique “band compression mechanism” of gradient elution can partially “reverse” it. For an overloaded band, the component concentration increases from the column inlet to the column outlet. The mobile phase with a higher organic phase proportion always reaches the low-concentration region of the band near the column inlet first, allowing them to continuously catch up with the high-concentration region near the column outlet, ultimately compressing the band and alleviating the previously tailing peak shape.
 Previous:
Previous: Back to List
Back to List